
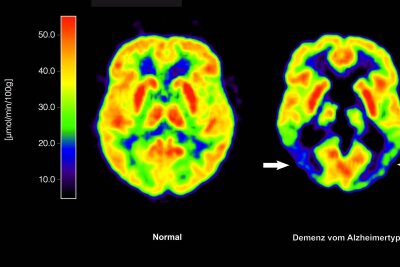
Allein in Deutschland sind etwa eine Million Menschen von Alzheimer betroffen. Die europäische Arzneimittel-Behörde EMA hat für die EU nun erstmals grünes Licht für eine Alzheimer-Therapie gegeben, die auf zugrundeliegende Krankheitsprozesse abzielt. Sie empfiehlt die Zulassung des Antikörpers Lecanemab zur Behandlung von leichter kognitiver Beeinträchtigung (Gedächtnis- und Denkstörungen) oder leichter Demenz in einem frühen Stadium der Alzheimer-Krankheit.
Warum ist die Entscheidung so besonders?
Bisherige Alzheimer-Therapien behandeln nur Symptome der Krankheit, nicht ursächliche Prozesse im Gehirn. Das ist bei Lecanemab anders: Der Antikörper richtet sich gegen Amyloid-Ablagerungen im Gehirn und soll dadurch den Verlauf der Krankheit verlangsamen. Um Heilung oder Verbesserung geht es allerdings auch bei diesem Wirkstoff nicht, ein solches Mittel ist weiterhin nicht in Sicht.
Hauptmaßstab für die Wirksamkeit war die Veränderung der kognitiven und funktionellen Symptome nach 18 Monaten, die anhand einer Demenzbewertungsskala gemessen wurde. Die Skala reicht von 0 bis 18, wobei höhere Punktzahlen eine stärkere Beeinträchtigung anzeigen. Mit Lecanemab behandelte Patienten wiesen nach 18 Monaten im Mittel einen etwas geringeren Anstieg des Wertes auf (1,22 gegenüber 1,75). Das deute auf einen langsameren kognitiven Abbau hin, teilte die EMA mit.
-
 Bisherige Alzheimer-Therapien behandeln nur Symptome der Krankheit, nicht ursächliche Prozesse im Gehirn. (Archivbild) Foto: Daniel Naupold/dpa
Bisherige Alzheimer-Therapien behandeln nur Symptome der Krankheit, nicht ursächliche Prozesse im Gehirn. (Archivbild) Foto: Daniel Naupold/dpa
Warum können nicht alle Alzheimer-Patienten Lecanemab bekommen?
Haben die Amyloid-Plaques schon irreversible Schäden im Gehirn angerichtet, nützt ihre Entfernung nichts mehr. Als frühe Alzheimer-Phase sind Johannes Levin vom Deutschen Zentrum für Neurodegenerative Erkrankungen (DZNE) zufolge die ersten drei Jahre zu werten. Das betreffe in Deutschland aktuell vermutlich mindestens 250.000 Menschen. In dieser Frühphase kommt ein Erkrankter noch sehr gut allein klar, merkt aber zunehmend, dass sein Gedächtnis nachlässt.
Bei der EMA-Empfehlung gibt es allerdings noch eine weitere Einschränkung: Das Mittel solle nur für Alzheimer-Patienten verwendet werden, die nur eine oder keine Kopie von ApoE4, einer bestimmten Form des Gens für das Protein Apolipoprotein E, haben. Bei ihnen ist die Wahrscheinlichkeit für bestimmte schwerwiegende Nebenwirkungen - Schwellungen und Blutungen im Gehirn - demnach geringer als bei Menschen mit zwei ApoE4-Kopien.
Menschen mit nur einer oder keiner ApoE4-Kopie machten je nach Region etwa zwei Drittel bis 80 Prozent der Alzheimer-Patienten aus, erklärte Gabor Petzold, Direktor der Klinischen Forschung am DZNE. In Deutschland seien es ungefähr 80 Prozent.
Hinzu kommen weitere einschränkende Voraussetzungen - insgesamt kommt Experten zufolge nur ein kleiner Bruchteil der Alzheimer-Erkrankten für eine Antikörpertherapie infrage.
Geht es jetzt direkt los mit solchen Behandlungen?
Nein. Zu bedenken sei, dass erst noch einige Schritte bis zu einem Einsatz in Deutschland anstünden, sagte Petzold: Die Zulassung durch die EU-Kommission stehe noch aus, zudem sei der Hersteller zum Beispiel verpflichtet worden, ausführliche Handreichungen und Schulungen unter anderem für Ärzte auszuarbeiten und ein Beobachtungsregister anzulegen.
Es werde noch einige Monate dauern, bis das Mittel wirklich eingesetzt werden könne. Bei Patienten müsse Alzheimer wiederum erst gesichert durch Biomarker-Tests nachgewiesen sein, gefolgt von einem genetischen Test. Infrage komme die Therapie zudem wirklich nur für Betroffene mit einer Vorstufe oder einem sehr frühen Stadium der Erkrankung.
In den nächsten Tagen sei aber bereits mit sehr vielen Anfragen von Betroffenen und Angehörigen bei Hausärzten, Alzheimer-Zentren und Gedächtnissprechstunden zu rechnen, sagte Petzold. Levin befürchtet einen Ansturm Zehntausender Menschen schon bei kleinsten Vergesslichkeiten. Für die Diagnosezentren wäre eine solche Flut von Abklärungen kaum zu stemmen, sagte er.
Der Neurologe Özgür Onur von der Uniklinik Köln geht zudem davon aus, dass er nur verhältnismäßig wenig Erkrankte pro Jahr mit der neuen Therapie behandeln kann, da die häufigen Gaben eine große Herausforderung darstellen. "Ich gehe bei uns in Köln von 50 bis 100 Patienten aus, die wir pro Jahr behandeln können. Und wir sind ein großes Zentrum."
Hatte die EMA Lecanemab nicht eigentlich schon abgelehnt?
Ja. Im Juli hatte die EU-Arzneimittelbehörde noch entschieden, dass das Risiko schwerer Nebenwirkungen des Antikörpers höher zu bewerten sei als die erwartete positive Wirkung. Das Unternehmen Eisai hatte daraufhin eine zweite Prüfung beantragt.
Dabei kam der Humanarzneimittelausschuss (CHMP) der EMA nun zu dem Schluss, dass in der begrenzten Population, die bei der erneuten Prüfung untersucht wurde, der Nutzen von Lecanemab bei der Verlangsamung des Fortschreitens der Krankheitssymptome größer ist als die Risiken. Bei der ersten Prüfung waren noch keine Untergruppenanalysen berücksichtigt worden, sondern alle Patienten.
Die für die Zulassung zuständige EU-Kommission folgt gewöhnlich dem Votum der Behörde. Hersteller von Lecanemab sind die Pharmaunternehmen Eisai (Japan) und Biogen (USA).
Warum der Bezug auf eine Untergruppe?
Bei den mit Lecanemab behandelten Patienten mit nur einer oder keiner ApoE4-Kopie traten der EMA zufolge bei 8,9 Prozent Ödeme im Gehirn auf, im Mittel aller Patienten bei 12,6 Prozent. Mikroblutungen gab es bei 12,9 Prozent der Patienten mit nur einer oder keiner ApoE4-Kopie, verglichen mit 16,9 Prozent der breiteren Population. Bei den Patienten mit nur einer oder keiner ApoE4-Kopie, die mit Placebo (einer Scheinbehandlung) behandelt wurden, lagen die Werte für Schwellungen bei 1,3 Prozent und für Blutungen bei 6,8 Prozent, wie es von der EMA hieß.
-
 Das Spielen von Memory soll gegen den Gehirnabbau wirken. Foto: Sven Hoppe/dpa
Das Spielen von Memory soll gegen den Gehirnabbau wirken. Foto: Sven Hoppe/dpa
Wie gefährlich sind solche Ödeme und Mikroblutungen?
Die erfassten Schwellungen und Mikroblutungen im Gehirn blieben überwiegend ohne Symptome und wurden zumeist erst durch bildgebende Verfahren wie Magnetresonanztomographie (MRT) bemerkt. Insbesondere bei wiederholtem Auftreten drohen jedoch eine verminderte Gehirnleistung oder Koordinationsschwierigkeiten. Mikroblutungen gelten zudem als Risikofaktor für größere, potenziell lebensbedrohliche Hirnblutungen.
Die EMA betont darum in ihrer Stellungnahme, dass es zwingend Maßnahmen zur Risikominimierung geben müsse. Vor Beginn der Behandlung und vor der 5., 7. und 14. Lecanemab-Dosis müssen bei den Patienten demnach MRT-Scans durchgeführt werden, zusätzliche Scans bei Warnzeichen wie Kopfschmerzen, Sehstörungen und Schwindel. Auch die Behandlung selbst ist aufwendig: Lecanemab wird als intravenöse Infusion alle zwei Wochen verabreicht.
Ist Lecanemab das einzige ursächlich wirkende Mittel?
Nein. Der Antikörper Aducanumab, entwickelt vom US-Biotech-Unternehmen Biogen, wurde von der EMA Ende 2021 nicht zur Zulassung empfohlen: Der vermeintliche klinische Effekt des Medikaments sei fraglich. Ein weiterer Zulassungsantrag wurde vom US-Pharma-Konzern Eli Lilly für den Wirkstoff Donanemab gestellt. Dieses Verfahren läuft noch.
Die US-Arzneimittelbehörde FDA ließ Aducanumab 2021 zur Alzheimer-Therapie zu, Biogen stoppte die Produktion Anfang des Jahres allerdings wieder. Anfang 2023 kam in den USA Lecanemab auf dem Markt. Donanemab wurde von der FDA am 2. Juli dieses Jahres zugelassen. Alle drei Antikörper haben einen ähnlichen Wirkmechanismus.
Gibt es Bedenken?
Im Fachjournal "The BMJ" äußerten Experten kürzlich Kritik an den FDA-Entscheidungen. Die Medikamente zeigten nur eine unmerkliche Verlangsamung der Demenz, dagegen jedoch schwerwiegende unerwünschte Nebenwirkungen, den Tod eingeschlossen, heißt es. Fragwürdig seien auch die finanziellen Verbindungen von Mitgliedern des FDA-Beratungsausschusses zu Pharma-Konzernen.
Kritik gibt es in "The BMJ" auch an den Aussagen der Hersteller, das Fortschreiten der Alzheimer-Krankheit werde deutlich verlangsamt - im Vergleich zu einer Placebotherapie je nach Teilgruppe um bis zu 35 Prozent. "Das ist eine irreführende Aussage", wird der Neurologe Alberto Espay von der Universität von Cincinnati speziell zu den Donanemab-Daten zitiert. "Das ist ein relativer Unterschied, der einen sehr kleinen absoluten Unterschied in eine Zahl verwandelt, die beeindruckend erscheint."
Fraglich ist, wie alltagsrelevant die messbare leichte Verzögerung des Krankheitsverlaufs überhaupt ist. "Sobald das Vollbild einer Alzheimer-Erkrankung vorliegt, sind die statistisch beschriebenen Effekte für den Patienten und sein Umfeld zumeist nicht mehr wahrnehmbar", sagte Walter Schulz-Schaeffer vom Universitätsklinikum des Saarlandes in Homburg. "Dem müssen die Nebenwirkungen des Medikaments entgegengesetzt werden."
Erwähnt wird im Fachjournal "The BMJ" zudem die in den Studien zu den Wirkstoffen gemachte Beobachtung, dass die Anti-Amyloid-Medikamente das Gehirn merklich schrumpfen lassen. Was es damit auf sich hat, ist bisher noch vollkommen unklar.

 Euer News-Tipp an die Redaktion
Euer News-Tipp an die Redaktion